Fully Automated Peptide Desalting for Liquid Chromatography–Tandem Mass Spectrometry Analysis Using Beckman Coulter Biomek i7 Hybrid Workstation
Casey W Coutelin Johnson1 , Qin Fu1 , Jennifer Van Eyk1 , Francis O Enane2, Michael Kowalski2, Bhagya Wijayawardena2
1 Advanced Clinical Biosystems Institute, Heart Institute, Cedars Sinai Medical Center. Los Angeles, CA
2 Beckman Coulter Life Sciences, Indianapolis IN
Abstract
Liquid chromatography-tandem mass spectrometry (LCMS/MS) is expected to play larger roles in quantitative proteomic research. Preparation of protein samples for LCMS/MS, however, is a multi-step labor intensive process involving protein denaturation, reduction, alkylation, protease digestion and peptide cleanup. These manual steps contribute to a significant analyst hands-on time yet reduces result turnaround and analyses times. Moreover, the labor-intensive steps contribute to a high rate of human errors, and in turn affect accuracy and reproducibility across individuals and laboratories, especially in high-throughput settings. This application note describes a completely hands-free automated procedure of the peptide desalting process needed for the peptide clean-up sample preparation steps of LCMS/ MS. Building from previous description of the automated trypsin digestion steps, current presentation allows for a completely hands-free LCMS/MS sample preparation protocols resulting in rigorous and reproducible high-throughput proteomic analysis. The automated procedure reduces hands-on time and demonstrated strong protein coverage resulting in high reproducibility in the results. This automation thus provides opportunities for high-throughput proteomics research that can result in wide characterization of protein-protein interaction research, global proteomic analysis and biomarker discovery in large sample size settings.
Introduction
Liquid chromatography and mass spectrometry (LCMS/MS) methods are commonly used in proteomic studies designed to cover various research backgrounds including but not limited to protein-protein interactions, global proteomic analyses and biomarker discovery1. Quantitative LCMS/MS provides researchers with the ability to quantitate and generate differential and relative amounts of proteins across biological samples such as patient samples or treated/untreated cells and tissues revealing biomarkers that generate insight into pathophysiology. Such analysis yields opportunities for global protein discovery that identifies unique biomarker candidates and verification and validation to confirm biomarker performance across varying sample sizes2.
Proteins analyzed can be derived from complex biological samples ranging from bodily fluids, biopsies, or cultured cells. Sample preparation is a crucial process in the proteomics workflow as the quality and reproducibility of the steps involved highly determines the separation and identification capabilities of mass spectrometers. However, samples must undergo a series of labor-intensive multi-step purification procedures that include protein solubilization and denaturation, disulfide bond reduction and cysteine blocking to ensure consistent cysteine masses, digestion of proteins into peptides with a site-specific protease cleavage, and cleanup to remove salts and denaturing agents2,3. Proteins undergo reduction and alkylation to open disulfide bonds as well as blockage of newly forming and preexisting cysteine side-chain residues4-6. Isolated proteins are then cleaved into smaller peptides using protease cleavage/ digestion by enzymatic cleavage such as trypsin digestion. This produces multiple small proteotypic peptides, that are building blocks of the corresponding protein chain, and that are easier to analyze by the LCMS/MS methods. Since MS measures charged ions, salts such as sodium and phosphate need to be removed to minimize their detection by MS. This can be achieved by desalting where buffer exchange or small molecule removal prevents interference with downstream analysis. While reproducibility in the sample preparation procedures is important to successful proteomic studies, there is a lack of high-throughput, reproducible, time and cost-effective assays to enhance large-scale LCMS/MS sample processing.
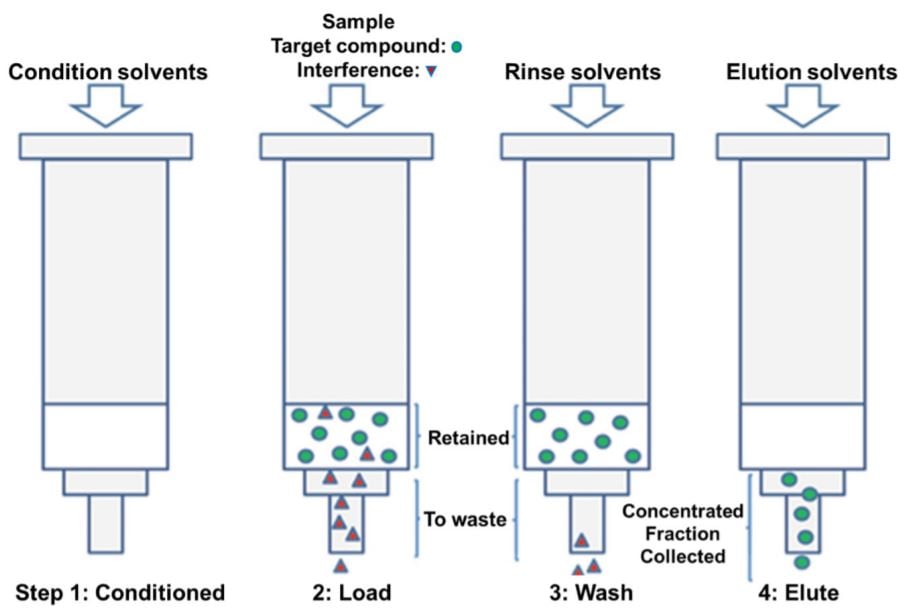
Figure 1. Sample extraction and purification steps. (A) Solid phase extraction. The steps used in the phase extraction included conditioning with methanol (MeOH) and 0.1% Formic Acid (FA), then Loading sample diluted with acid (2% H3PO4, 0.5% FA), rinsing with 0.1% FA, and Elution with 50% Acetonitrile (ACN) in 0.1% FA. This is then followed by pumping to dryness.
Developing automated sample preparation procedures that are time efficient, highly reproducible, and hands-free can enhance the LCMS/MS research by increasing the level of throughput and ultimately decrease the time from biomarker discovery to biomarker validation. Previously, we automated a protein digestion workflow leading to increased throughput and reproducibility2. Here we attempt to automate the entire LCMS/MS sample preparation workflow by automating the peptide desalting using the Beckman Coulter Biomek i7 Hybrid workstation.
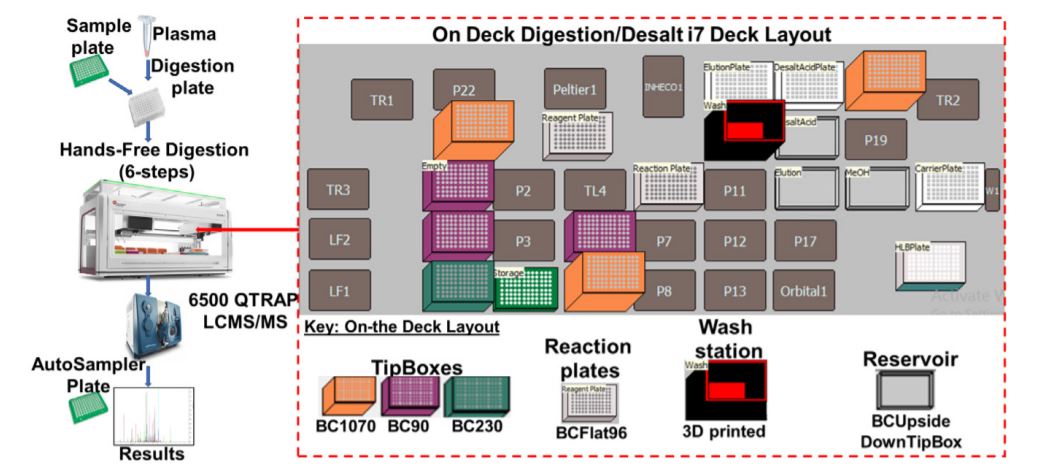
Figure 2. Robotic digestion and desalting steps on the Biomek i7 automated work station. Following the sample preparation, the sample plate is subjected to series procedures that collectively result in the sample digestion and desalting. The on-deck arrangement of equipment, reagents and resources needed for robotic automation and liquid handling is shown.
Methods
HLB plate conditioning
The Waters Oasis HLB 96-well plate (30 mg sorbent per well, 30 µm) was loaded into the Positive Pressure Apparatus (PPA) moving platform equipped to a vacuum powered drain. For a full plate, the 96 wells were initially conditioned using 1 mL methanol. This step was performed using the Span-8 pipetting arm due to methanol’s high volatility. An integrated Positive Pressure X-Well SPE Extractor (Amplius, Germany) was then used for Solid Phase Extraction. The bottom surface of the Positive Pressure X-Well SPE Extractor arm is cushioned with rubber that contains 96 holes correlating with the HLB plate. Using compressed air, pressure was applied to the top of these wells in a slowly increasing gradient of pressure so that a dropwise flowrate was maintained through every well. The process continues long enough for each well to be completely emptied of solvent (Figure 1A), which is then removed from the platform via the vacuum power drain.
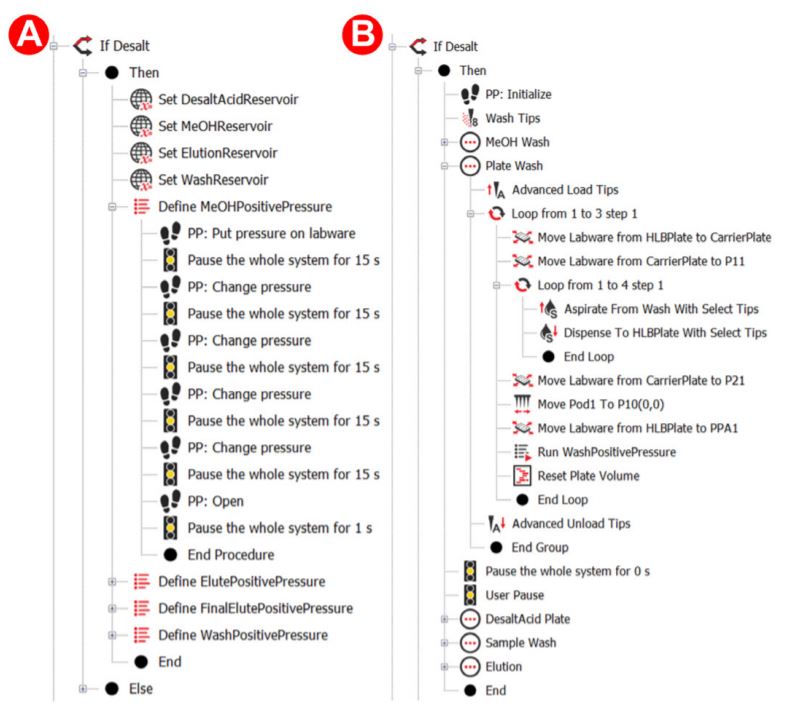
Figure 3. Biomek methods for desalting
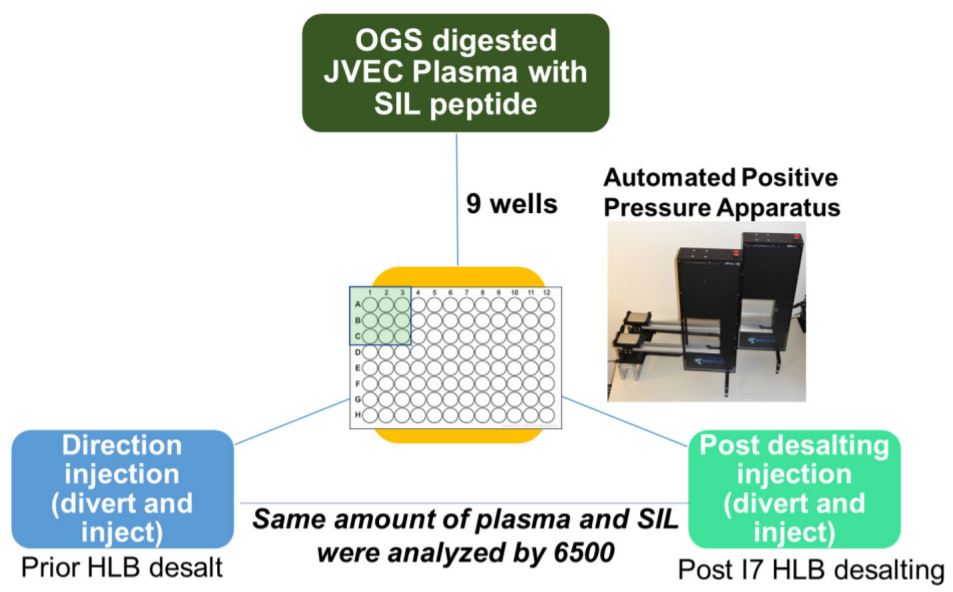
Figure 4. Biomek methods for desalt Reproducibility of i7 automated HLB Desalting Workflow. Samples were performed in 9 wells on three different days. Preparation on Biomek i7 series for quantitative proteomics.
HLB plate washing and peptide eluting
The HLB plate was further conditioned with 1 mL of 0.1% formic acid (FA) in HPLC grade water three times. This was accomplished by placing the HLB plate on top of a ‘carrier plate’, which is moved closer to the 96 well pipetting head. A 250 µL volume was then transferred four times by the 96 well plate. Using this ‘carrier plate’ method instead of the Span-8 method reduced this plate’s processing time from ~5 minutes to less than 2 minutes. The digested protein mixture was then diluted with an acid mixture and mixed, preparing the peptides to be loaded onto the HLB plate. When the PPA applies pressure to the loaded samples, the applied pressure gradient was even more slow and gradual, ensuring the dropwise flow and proper loading of samples onto the sorbent of the HLB plate. This was followed by three more washes of the loaded samples using 1 mL of 0.1% FA to remove impurities (Figure 2) using Biomek methods for the automation steps (Figure 3).
To achieve peptide elution in the final step, the HLB plate was loaded on top of a 96 well 2 mL deepwell plate. The 96 well pipetting head was then used to transfer a solution of 50% acetonitrile in 0.1% HPLC grade water onto the HLB plate. The PPA then applied a slow and gradual pressure gradient to the wells to allow the organic/aqueous solution to elute the desalted peptides off of the sorbent and into the deepwell plate below, completing the desalting process.
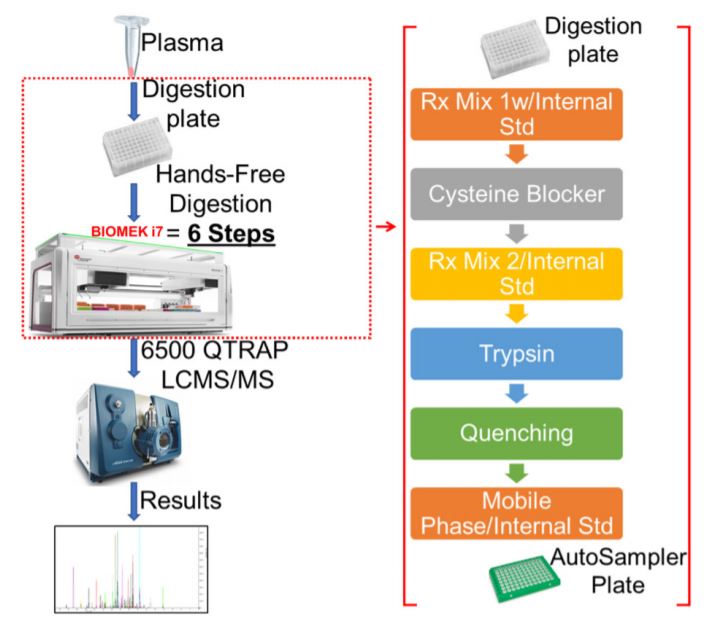
Figure 5. Schematic for MRM Proteomics Workflow using i7. Step by step workflow for automated proteomics sample preparation on Biomek i7 for quantitative proteomics.
A large amount (~800 µL for 96 plate) of 0.1% FA is used in the entire process, so a custom piece of labware was designed to serve as a large reservoir (Figure 2). Using a 3D printer, the reservoir was printed using polylactic acid (PLA) filament. This printing technique and material used is permeable to water, so the reservoir was designed to accommodate a small polypropylene (PP) plastic liner. Automated methods were used for peptide digestion steps (Figure 5).
Results
Positive Pressure X-Well SPE Extractor was Integrated onto the Biomek i7 workstation as an alternative to manual desalting apparatuses. Desalting via solid phase extraction is a known technique of separating soluble macromolecules from smaller molecules (Figure 2). This allows for the removal of reagents and impurities from digested protein samples, yielding cleaner peptide mixtures. This is especially helpful, if not essential, when loading samples onto nanoflow liquid-chromatography (LC) instruments and highly sensitive mass spectrometers (MS).
The automated method used here was designed to be robust and reproducible (Figure 4), where samples were done in triplicate in 9 wells of a 96 well plate. We observed equal intensities in samples direction injection and without desalt as compared to HLB- desalted by PPA on the Biomek i7 workstation prepared samples (Figure 6A) suggesting that automation did not affect sample quality and peptide stability. Moreover, we also observed good sampleto-sample reproducibility (5-10% CV) (Figure 6B) obtained across 9 replicate samples. There was a high reproducibility where an average CV of 10-20% was observed (Figure 7A-B).
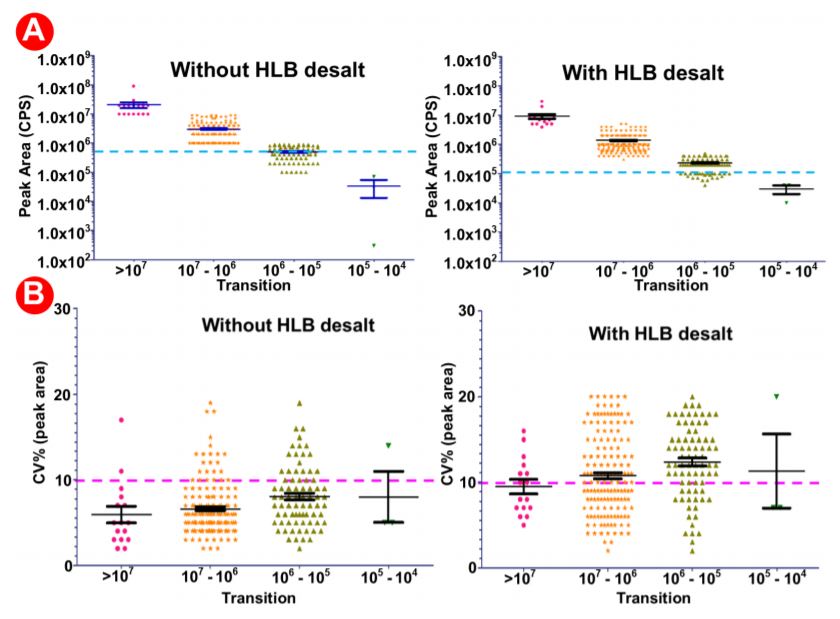
Figure 6. Analysis of data generated with/without HLB desalting and automated proteomic sample preparation workflow with a highly multiplexed SRM analysis. 9 digested plasma peptides aliquots were processed. The resulting peptides were analyzed by LCMS/ MS with a 30 min LC gradient and a scheduled SRM method targeting multiple proteins. (A) Average intensities in samples prepared without HLB (left) versus those prepared with HLB (right). (B) % CVs in 9 samples prepared without HLB (left) versus those prepared with HLB (right).
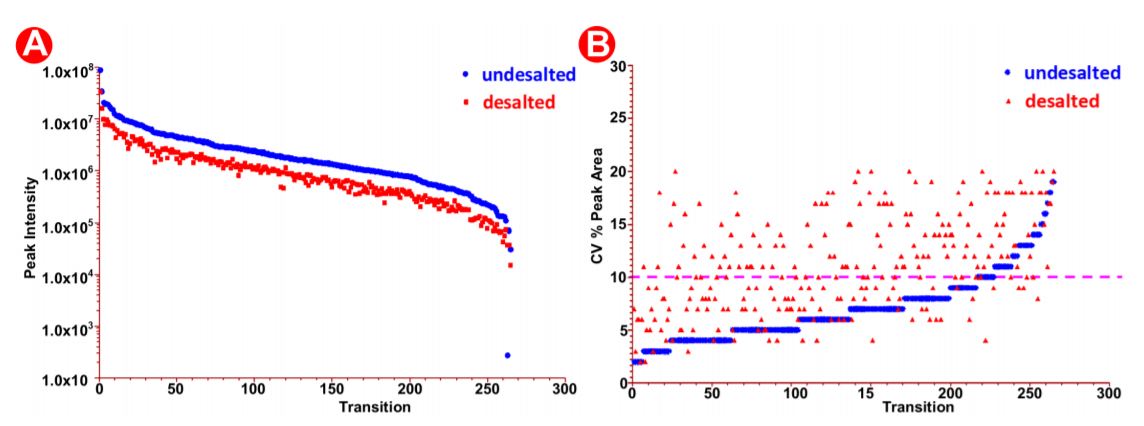
Figure 7. Peak area analysis in pre/post-HLB desalted samples. (A) Peak intensity in desalted versus undesalted samples. (B) SIL peak area CV% in the desalted versus undesalted samples.
Summary
In summary, this application note demonstrates a successful automation of the peptide desalting step in the LCMS/MS proteomics workflow. In addition to the previous automation of the protein digestion workflow2, we now have a fully automated sample preparation workflow for proteomics samples needed for LCMS/MS analysis using the Biomek i7 hybrid workstation. The results indicate a highly reproducible and quantitative datasets for peptides generated across all samples analyzed, suggesting a broad applicability of this method to high-throughput quantitative proteomics. The Biomek i7 series has a high deck capacity, with opportunity for multiple on-deck integrations to reduce the hands-on time.
References
- Sajic T, Liu Y, Aebersold R (2015) Using Data-Independent, High Resolution Mass Spectrometry in Protein Biomarker Research: Perspectives and Clinical Applications. Proteomics Clin Appl 9(3-4):307–321
- Fu Q, Kowalski MP, Mastali M, et al. Highly Reproducible Automated Proteomics Sample Preparation Workflow for Quantitative Mass Spectrometry. J Proteome Res. 2018;17(1):420-428. doi:10.1021/acs.jproteome.7b00623
- Nilsson J et al. (2013) LC-MS/MS characterization of O-glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J Proteome Res 12(2):573–584
- Rogers RS, Nightlinger NS, Livingston B, Campbell P, Bailey R, Balland A. Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics. mAbs. 2015;7:881-90.
- Mouchahoir T, Schiel JE. Development of an LC-MS/MS peptide mapping protocol for the NISTmAb. Anal Bioanal Chem. 2018;410:2111-26.
- Richardson J, Shah B, Xiao G, Bondarenko PV, Zhang Z. Automated in-solution protein digestion using a commonly available high-performance liquid chromatography autosampler. Anal Biochem. 2011;411:284-91.
AAG-7941APP09.20
