Effective Miniaturization of Illumina Nextera XT Library Prep for Multiplexed Whole Genome Sequencing and Microbiome Applications
Abstract
The cost of sequencing per nucleotide has been declining by orders of magnitude over the past ten years; however library preparation of samples for next-generation sequencing remains a costly component of the process. In synthetic biology, microbial whole genome sequencing is becoming a routine process to validate genome editing, deconvolute mutagenesis campaigns, confirm highthroughput DNA construction, and survey microbiome samples. Here we detail a method to miniaturize reaction volumes utilized in the Illumina Nextera XT DNA library kit, thereby reducing the overall cost per reaction while maintaining equivalent data to the standard protocol. We then tested this miniaturized method on a diverse variety of microbes to show that the method is effective across samples. Finally, we applied this method to a microbiome study and found that it maintains sample diversity, ratios, and identification.
Introduction
The cost of sequencing per nucleotide has been declining by orders of magnitude in the past ten years, due to continuous improvement in next-generation sequencing technologies. However, library preparation of samples remains a bottleneck for reducing sequencing costs. In synthetic biology, whole genome sequencing is becoming a routine process to validate genome editing, deconvolute mutagenesis campaigns, confirm high-throughput DNA construction, and survey microbiome samples. Current efforts to sequence small genomes (bacterial, fungal) using an Illumina Nextera XT-based workflow are constrained by the minimum working volumes needed for manual processing, and once processed, only a small fraction of the library generated is used for the sequencing run. By utilizing the Echo 525 Liquid Handler with the Nextera XT kit, we perform the reactions at a miniaturized scale that reduces excessive library generation, thus saving costs on expensive reagents. In addition, running smaller volume tagmentation reactions also reduces the amount of sample gDNA required. We demonstrate that the tagmentation and the amplification reactions can be scaled down 20-fold. These libraries were multiplexed on an Illumina MiSeq paired-end sequencing run and shown to produce equivalent quality metrics. The nanoliter-precision and speed of the Echo 525 Liquid Handler enables accurate miniaturization of Nextera XT library preparation and QC measures such as the Quant-iT Picogreen quantification assay and Agilent TapeStation 2200 fragment analysis, thus reducing cost and increasing throughput while providing high quality sequencing data. Furthermore, the Echo 525 Liquid Handler enabled us to normalize and pool our libraries for sequencing at a speed unrivaled by any tipbased liquid handler.
Methods and Discussions
Miniaturization of Tagmentation and Amplification Reactions
In an effort to address volumetric limitations with manual workflows, we undertook an experiment with the Echo 525 Liquid Handler to test a panel of reaction volumes with the aim of reducing cost per sample while maintaining sequencing quality. The E. coli K-12 genome was selected for this study, because of its widespread use in academic and industrial settings. In addition, its representative “small genome” size allows for a high degree of multiplexing conditions of library prep on one MiSeq run. All samples were run in three technical replicates. The Echo 525 Liquid Handler was used to transfer differing amounts of the same sample gDNA into a variety of tagmentation reaction volumes, then was also used to dispense the appropriate TD + ATM buffer volumes, except for the 25µL manual control. 55°C tagmentation incubation was performed in the ThermoFisher ProFlex thermocycler. A different PCR plate was used for each reaction volume size to account for differences in heating. To neutralize tagmentation, NT buffer was dispensed using the Echo 525 Liquid Handler. PCR amplification of tagmented libraries was performed at various volumes. Indexing primers and Nextera PCR Mix (NPM) were dispensed at varying volumes by the Echo 525 Liquid Handler. Amplification was run on a reaction volume basis following the Nextera XT PCR conditions. Libraries were then cleaned up manually utilizing SPRI bead cleanup at a ratio of 0.6x. Each library was then quantified via the Picogreen assay. The Echo 525 Liquid Handler was utilized in this assay to dispense the samples, as well as Picogreen reagents to a miniaturized 20µL reaction in a Greiner 384-well plate, and results were read on the BMG PHERAstar. Each library also underwent fragment size analysis via the TapeStation 2200. The Echo 525 Liquid Handler was used to dispense sample and reagent into the assay plate. Using a combination of quantification data and average fragment size data, we generated a normalization worklist. The Echo 525 Liquid Handler was then able to simultaneously normalize and pool the libraries together by transferring small, accurate, and precise amounts of libraries into one well. This pooled library was then quantified via Qubit, normalized to 4nM, denatured via sodium hydroxide, then loaded onto an Illumina MiSeq instrument for paired-end 300bp reads. Data were then analyzed in the Biomatters Geneious software, aligned to E. coli K-12 MG1655 reference genome from NCBI using the Geneious aligner. A summary of results is published in the table below.
| Input DNA | Tagmentation Volume | PCR Volume | Fragment Size | Coverage | Mean Q-Score | > Q30 | |
| Average | StDev | ||||||
| 1 ng | 25 µL | 25 µL | 419 bp | 9 | 4.5 | 33.7 | 82.70% |
| 1 ng | 25 µL | 25 µL | 410 bp | 8.5 | 4.6 | 33.5 | 81.80% |
| 0.5 ng | 12.5 µL | 6.25 µL | 372 bp | 8.4 | 4.6 | 34.4 | 85.50% |
| 0.25 ng | 6.25 µL | 6.25 µL | 378 bp | 9.4 | 5 | 33.9 | 83.70% |
| 0.05 ng | 1.25 µL | 2.5 µL | 335 bp | 9.7 | 5.5 | 34.1 | 84.40% |
TABLE 1: Individual library conditions and resulting coverage and quality
25 μL Tagmentation and 25 μL PCR
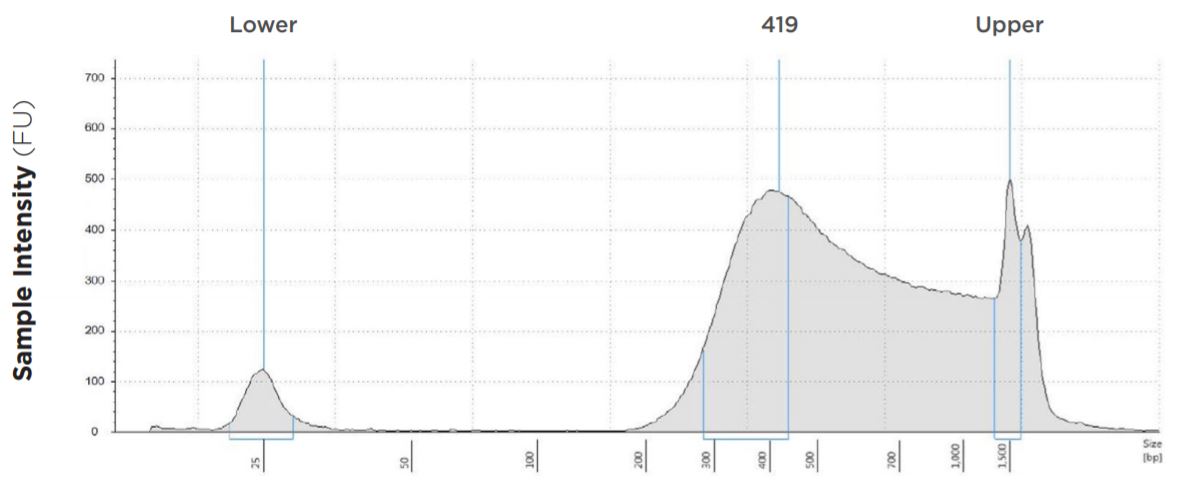
FIGURE 1A: TapeStation electropherograms of conditions tested, post magnetic bead cleanup. Size selection cutoff was performed using 0.6x SPRI beads. Average fragment length was 419 bp with a right tail, appropriate for 2x300 reads. Peak size dropoff reflects lesser amount of product amplified in a smaller volume.
25 μL Tagmentation and 2.5 μL PCR
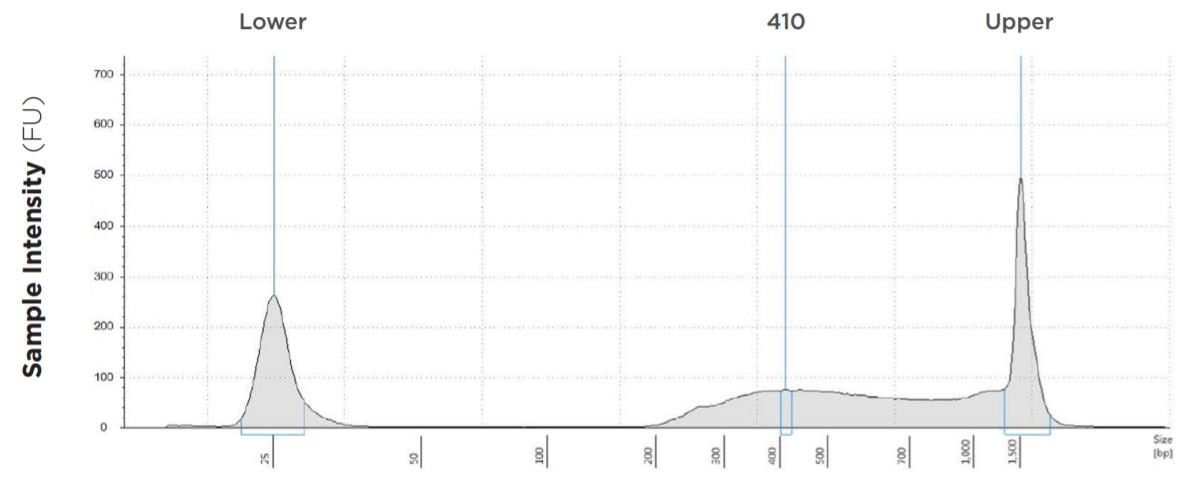
FIGURE 1B: TapeStation electropherograms of conditions tested, post magnetic bead cleanup. Size selection cutoff was performed using 0.6x SPRI beads. Average fragment length was 410 bp with a right tail, appropriate for 2x300 reads. Peak size dropoff reflects lesser amount of product amplified in a smaller volume.
12.5 μL Tagmentation and 6.25 μL PCR
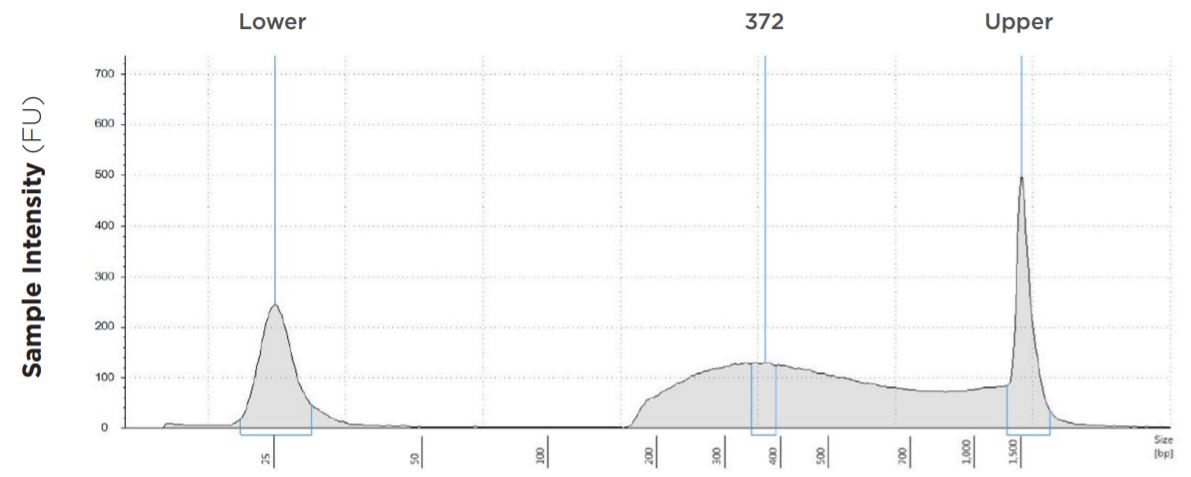
FIGURE 1C:TapeStation electropherograms of conditions tested, post magnetic bead cleanup. Size selection cutoff was performed using 0.6x SPRI beads. Average fragment length was 372 bp with a right tail, appropriate for 2x300 reads. Peak size dropoff reflects lesser amount of product amplified in a smaller volume.
6.25 μL Tagmentation and 6.25 μL PCR
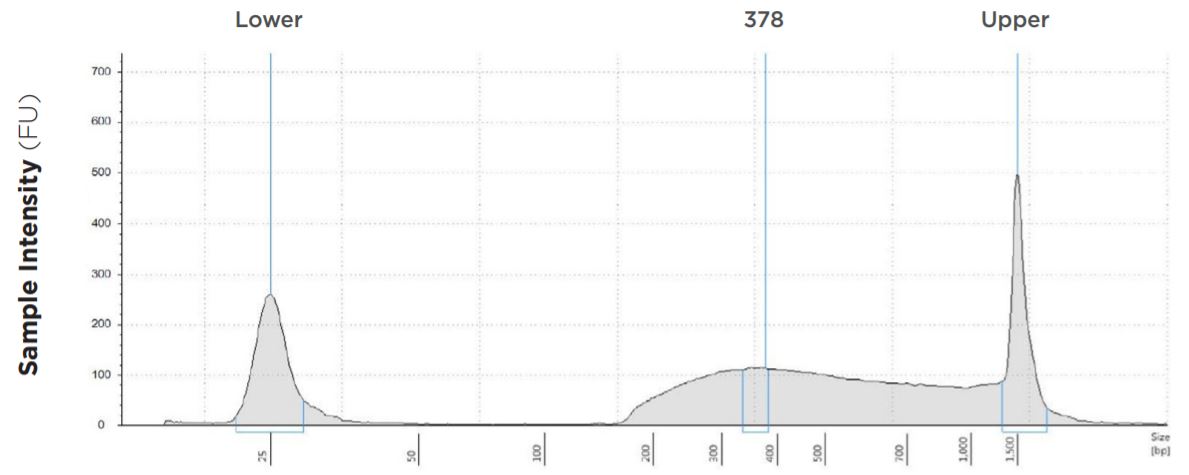
FIGURE 1D: TapeStation electropherograms of conditions tested, post magnetic bead cleanup. Size selection cutoff was performed using 0.6x SPRI beads. Average fragment length was 378 bp with a right tail, appropriate for 2x300 reads. Peak size dropoff reflects lesser amount of product amplified in a smaller volume.
1.25 μL Tagmentation and 2.5 μL PCR
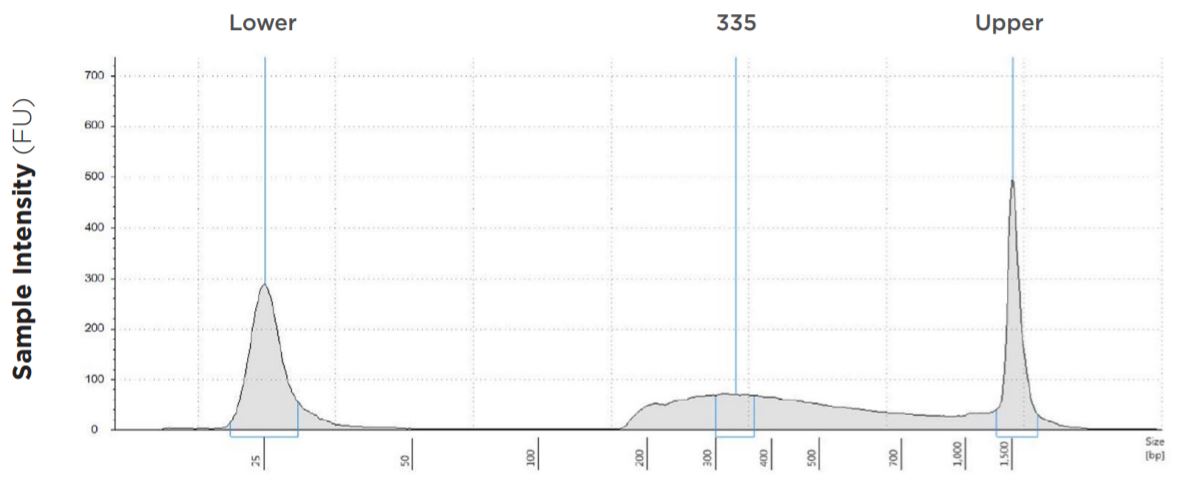
FIGURE 1E: TapeStation electropherograms of conditions tested, post magnetic bead cleanup. Size selection cutoff was performed using 0.6x SPRI beads. Average fragment length was 335 bp with a right tail, appropriate for 2x300 reads. Peak size dropoff reflects lesser amount of product amplified in a smaller volume.
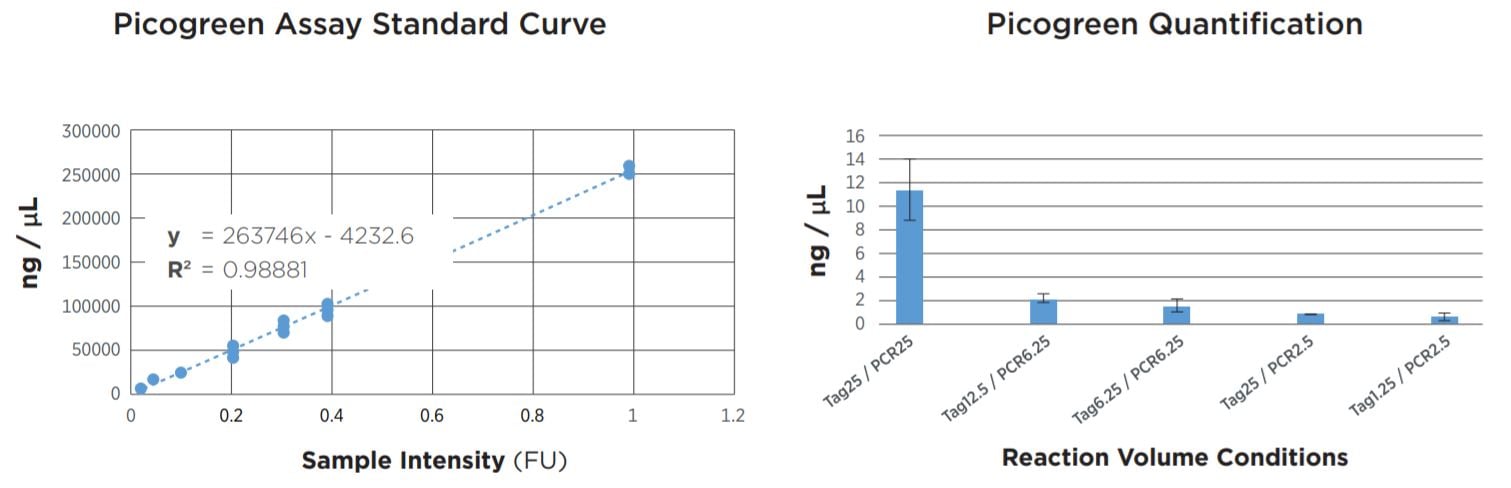
FIGURE 2: Picogreen quantification results. Standard curve was generated using lambda DNA from 25pg/µL to 1µg/µL. Results agree with TapeStation fragment analysis. Yield dropoff is expected due to reduced PCR volume, and is reproducible across technical replicates. Tag25/PCR25 was performed manually and shows greater variability.
Q-Scores per Cycle
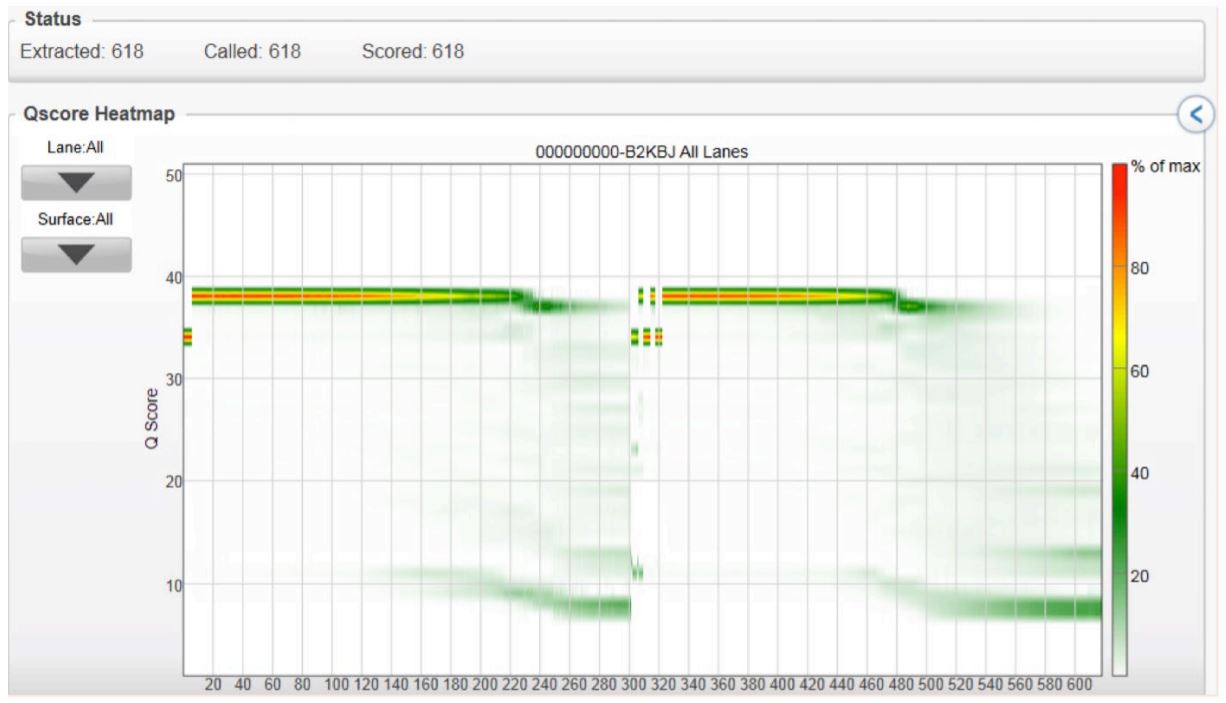
FIGURE 3: Quality read scores per cycle of the volume-multiplexed MiSeq run, demonstrating that various reaction volumes can be run together and produce quality scores similar to conventional 2x300 genomic runs.
Coverage for Alignments to RefSeq

FIGURE 4: Coverage graphs from Geneious alignments. From top to bottom: Tag25/PCR25, Tag25/PCR2.5, Tag12.5/ PCR6.25, Tag6.25/PCR6.25, and Tag1.25/PCR2.5. Coverage is highly equivalent regardless of reaction volumes used. E.coli K-12 gDNA from ATCC was aligned to E.coli K-12 MG1655 reference genome from NCBI.
During the course of the experiment, we discovered that fragmentation remains effective at miniaturized reaction volumes. While certainly less library is generated in a smaller volume amplification reaction and subsequent PCR cleanup, this can be mitigated by utilizing the Echo 525 Liquid Handler’s ability to rapidly normalize concentrations based on Picogreen quantification data. During library QC, we found that all volumes of tagmentation produced similar average fragment length and size distributions. After sequencing, we see that there is no significant effect to the coverage, standard deviation, and Q-Score when comparing the manual workflow with Echo Liquid Handler-enhanced miniaturized workflows. This allows for reduction of reaction volumes without compromising data, resulting in reagent cost and input DNA savings. This experiment gave us confidence to utilize 2.5µL/5µL tagmentation/PCR reaction volumes for the following production experiment. These volumes were chosen for their ease of compatibility with our laboratory equipment and wares.
High Throughput Library Preparation and Multiplexing Whole Genome Sequencing
To represent an academic/industrial production situation, we chose nine diverse and commonly studied organisms and one microbiome sample to run together on a MiSeq. Based on the genome sizes and available output of a MiSeq v3 Reagent Kit, we were comfortable multiplexing these samples and producing coverage with strong statistical confidence. We wanted to test the robustness of the miniaturized workflow with a variety of genome sizes, GC content, and genome organizations. Additionally, the microbiome sample contained 10 organisms commonly tested for in that application. We sought to determine if organism and strain identification was possible at this scale, and if organism frequencies were maintained throughout the process. The sample gDNA for each organism was first manually normalized to a concentration of 5ng/µL. Then, the library preparation procedure was performed as described in the above experiment, now utilizing 2.5µL/5µL tagmentation/PCR reaction volumes. Libraries were then cleaned up manually utilizing SPRI bead cleanup at a ratio of 0.6x, quantified via Picogreen assay, and analyzed via TapeStation 2200. Using a combination of quantification data and average fragment size data, we generated a normalization worklist. The Echo 525 Liquid Handler was then able to simultaneously normalize and pool the libraries together by transferring small, accurate, and precise amounts of libraries into one well. This pooled library was then quantified via Qubit, normalized to 4nM, denatured via sodium hydroxide, then loaded onto an Illumina MiSeq instrument for paired-end 300bp reads. Each sample was aligned to its respective reference genome or close relative from NCBI, and sequencing data were calculated using Biomatters Geneious software and the Geneious aligner.
| Species | Genome Size | MiSeq Index Representation | Alignment to RefSeq | Coverage | StDev | Confidence Score | Fragment Size |
| Salmonella enterica | 4.70 Mbp | 5.94% | 98.3% | 53.8x | 23.6 | 34.7 | 438 bp |
| Escherichia coli | 4.65 Mbp | 1.20% | 99.40% | 12.5x | 6 | 33.2 | 504 bp |
| Pseudomonas aeruginosa | 6.34 Mbp | 1.01% | 93.80% | 7.9x | 4.8 | 31.1 | 481 bp |
| Saccharomyces cerevisiae | 12.16 Mbp | 9.36% | 99.80% | 34.3x | 21.9 | 34.7 | 420 bp |
| Pichia pastoris | 9.59 Mbp | 1.59% | 99.60% | 9.9x | 7.5 | 34.2 | 436 bp |
| Cryptococcus neoformans | 18.91 Mbp | 3.45% | 98.40% | 10.4x | 5.6 | 33.7 | 512 bp |
| Listeria monocytogenes | 2.95 Mbp | 5.51% | 91.60% | 80.1x | 47.7 | 35.3 | 401 bp |
| Staphylococcus aureus | 2.8 Mbp | 4.30% | 99.99% | 83.3x | 40.3 | 35.3 | 444 bp |
| Enterococcus faecalis | 3.36 Mbp | 3.55% | 99.94% | 61.1x | 31.1 | 34.8 | 457 bp |
| ZymoBIOMICS | 6.07% | 396 bp |
TABLE 2: Summary data for each sample that was included on a multiplexed 2x300 MiSeq run.
Salmonella enterica
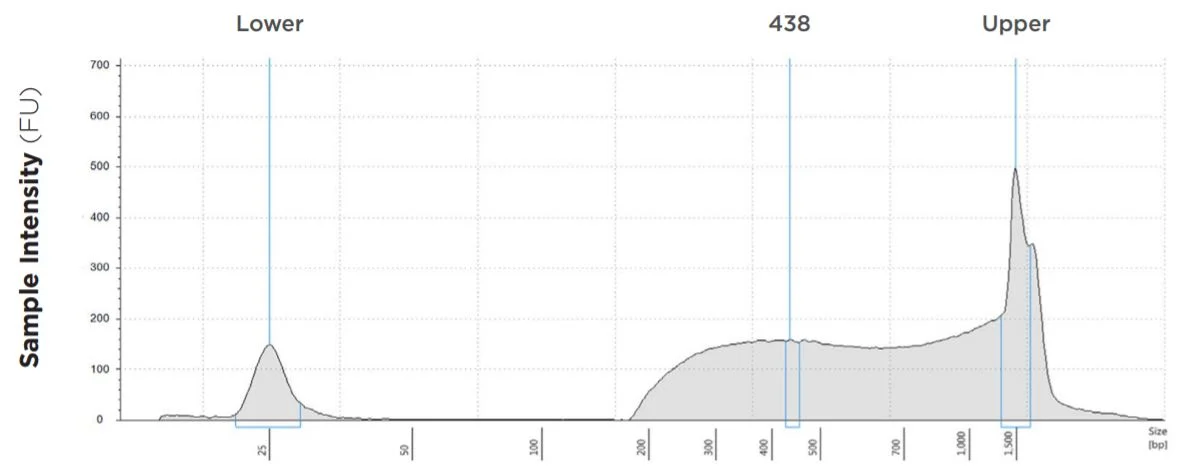
FIGURE 5: TapeStation electropherogram after library prep and magnetic bead cleanup of Salmonella enterica, a representative sample. Size selection cutoff was performed using 0.6x SPRI beads. Average fragment length is about 438 bp with a right tail, appropriate for 2x300 reads.
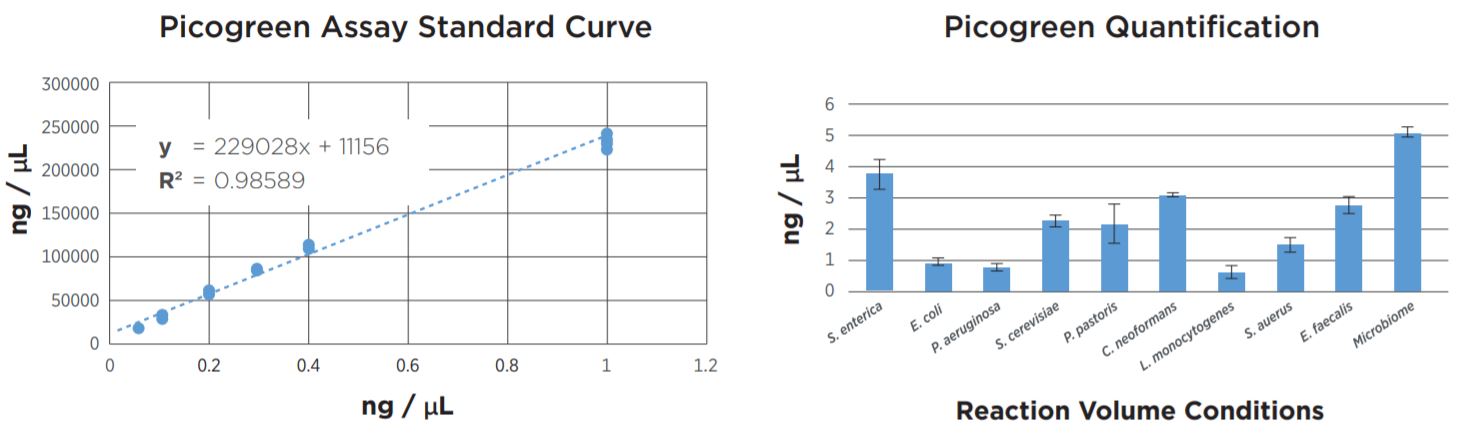
FIGURE 6: Picogreen quantification data of the samples after library prep and SPRI bead cleanup using the Echo 525 Liquid Handler to set up the assay. While the yield per sample varies, the Echo 525 was able to normalize and pool the samples together in less than one minute.
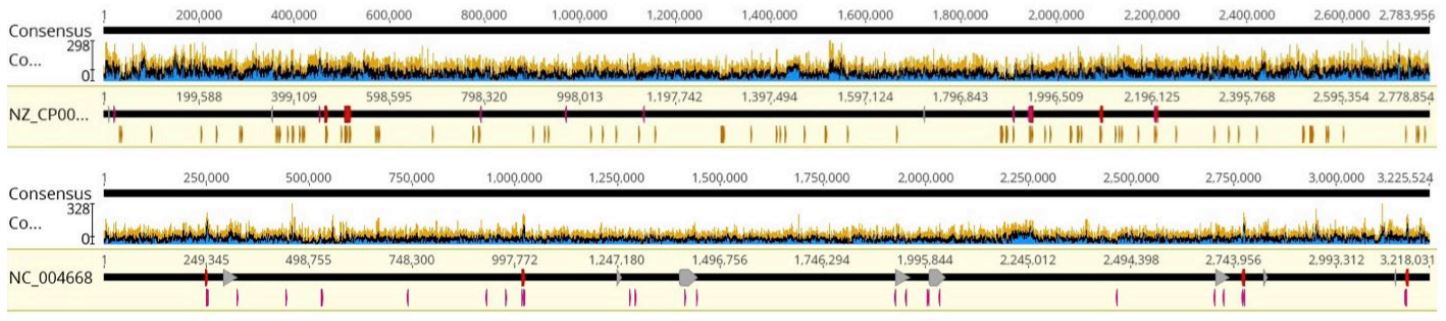
FIGURE 7: Coverage graphs of a) Staphylococcus aureus and b) Enterococcus faecalis, representative samples.
For three of the samples, the specific strains/serovar sequenced did not have a NCBI reference sequence available; a close strain relative was used in these cases. This explains the lower values in mapping reads to the reference (Alignment to RefSeq). These samples are Salmonella enterica, Pseudomonas aeruginosa, and Listeria monocytogenes. Otherwise, nearly all the reads are mapping to their respective reference, the genome is well-covered, and confidence scores are high. This holds true across the various genome sizes, GC content, and genome organization. The fluctuation in coverage between samples is expected. Coverage is a function of both the number of reads allocated to the sample (MiSeq Index Representation), and the number of base pairs needing to be covered (genome size). More optimization and normalization can be done to further unify the coverage across samples. All in all, we show that the Echo Liquid Handler-enhanced miniaturized workflow can produce high-quality whole genome sequencing data across various microbes while reducing reagent costs and input.
Microbiome
The goal of the microbiome sample in the previous experiment was to determine whether an Echo 525 Liquid Handler-enhanced library preparation workflow would preserve the ability to identify organisms as well as preserve the ratio in which they are initially supplied. To test this, we obtained a microbial community DNA standard from Zymo Research, and processed it in tandem with the other samples during the previous experiment. The microbiome sample was barcoded using one pair of indices. After de-multiplexing, total read count was obtained from this sample, and reads were aligned to each of the organisms that were purported to be included in the microbiome using the Geneious aligner in Geneious software by Biomatters. Successful alignments were calculated as a ratio of the whole, then compared to the given gDNA abundances.
| Source | Organism | Reported GC | Measured GC | gDNA Abun. (%) | Reads | Measured Abun. (%) |
| ZymoBiomics D6305 | Pseudomonas aeruginosa | 66 | 67 | 12 | 209,673 | 14 |
| Escherichia coli | 57 | 53 | 12 | 242,562 | 16 | |
| Salmonella enterica | 52 | 53 | 12 | 297,457 | 20 | |
| Lactobacillus fermentum | 53 | 53 | 12 | 162,806 | 11 | |
| Enterococcus faecalis | 38 | 41 | 12 | 113,919 | 8 | |
| Staphylacoccus aureus | 33 | 38 | 12 | 126,185 | 8 | |
| Listeria monocytogenes | 38 | 41 | 12 | 124,416 | 8 | |
| Bacillus subtilis | 44 | 46 | 12 | 179,820 | 12 | |
| Saccharomyces cerevisiae | 38 | 39 | 2 | 27,137 | 2 | |
| Cryptococcus neoformans | 48 | 48 | 2 | 46,946 | 3 | |
| 1,487,546 | ||||||
TABLE 3: Reads successfully aligned compared to their given values.
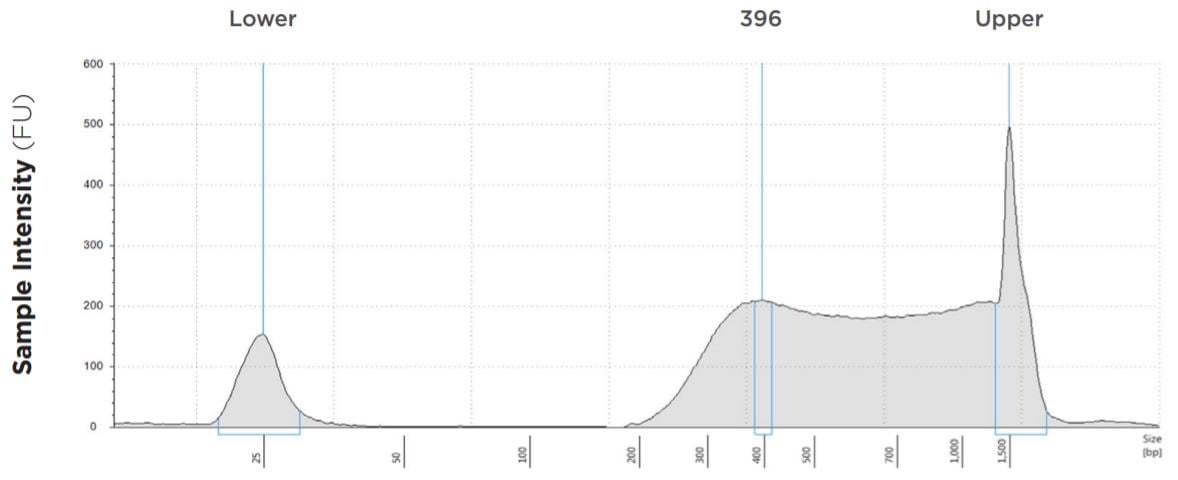
FIGURE 8: TapeStation electropherogram after library prep and magnetic bead cleanup. Size selection cutoff was performed using 0.6x SPRI beads. Average fragment length is about 396bp with a right tail, appropriate for 2x300 reads.
We have found that after miniaturized library preparation and sequencing of the microbiome sample, reads are mapped to their respective organism at ratios reflecting their initial abundance. For the purposes of microbiome identification and population survey, the miniaturized library preparation does not introduce significant biases despite the typically GC-biased enzymatic reactions. This method can be used for microbiome studies and metagenomic surveys to probe for diversity and population of organisms and genes of interest.
Conclusion
We have detailed a method here for miniaturizing reaction volumes utilized in the protocol for the Illumina Nextera XT DNA library prep kit and we applied this to a variety of small genome samples as well as a microbiome sample. We also described protocols for associated QC measures such as the Quant-iT Picogreen assay and Agilent TapeStation 2200 fragment analysis. Finally, we highlighted the impact of utilizing the Echo 525 Liquid Handler in the normalization and pooling of libraries.
The ability to deliver volumes as low as 25nL affords an obvious reduction in reagent volumes leading to reduced costs for the researcher and higher throughput possible with the same quantity of supplied reagents. In this case, we saw a 20-fold reduction in the volumes for tagmentation and PCR reactions with no significant impact to the coverage, standard deviations, and confidence scores when compared to the manual protocol reaction volumes.
In addition to reduced costs, the flexibility and any well to any well capabilities of the Echo series of liquid handlers also enables easy assay optimization, allowing the researcher to test a wide range of conditions often from a single source plate. With the miniaturization of tagmentation and amplification protocols, we were able to easily carry out a series of experiments with a defined range of volumes that led to the determination of a final optimized 2.5 µL/5 µL tagmentation/PCR reaction volume.
The highly accurate and precise transfer of 25nL increments enabled normalization and pooling of samples in a single step, thus affording time savings that are impossible to achieve with standard tipbased liquid handling.
The Echo 525 Liquid Handler enhanced miniaturization of library preparation across a diverse range of organisms, providing consistent, high-quality results for whole genome sequencing despite varying phyla, GC content, genome size, and genome organization. This method can also be applied to microbiome and metagenomic applications, as the miniaturized process maintains sample diversity, ratios, and identification.
| Equipment | Manufacturer |
| Echo 525 Liquid Handler | Beckman Coulter Life Sciences |
| Allegra X-14 Centrifuge | Beckman Coulter Life Sciences |
| MixMate | Eppendorf |
| Qubit | ThermoFisher |
| TapeStation 2200 | Agilent |
| BMG PHERAstar | BMG Labtech |
| ProFlex PCR System | ThermoFisher |
| 384-well Post Magnet Plate | Alpaqua |
| MiSeq Sequencing Instrument | Illumina |
| Reagents | Manufacturer | Part Number |
| Nextera XT DNA 96-Sample Prep Kit | Illumina | FC-131-1096 |
| Nextera XT Index Kit v2 Set A | Illumina | FC-131-2001 |
| PhiX Control v3 | Illumina | FC-110-3001 |
| TapeStation D1000 HS Kit | Agilent | #5067-5584, #5067-5585 |
| Qubit dsDNA HS Assay Kit | ThermoFisher | #Q32851 |
| Quant-IT Picogreen dsDNA Assay Kit | ThermoFisher | #P11496 |
| Agencourt AMPure Beads | Beckman Coulter Life Sciences | #A63881 |
| 200 Proof Ethanol | Sigma Aldrich | #E7023 |
| MiSeq Reagent Kit v3 (600-cycle) | Illumina | MS-102-3003 |
| Salmonella enterica | ATCC | BAA-1740D-5 |
| Escherichia coli | ATCC | 10798D-5 |
| Pseudomonas aeruginosa | ATCC | 9027D-5 |
| Saccharomyces cerevisiae | ATCC | 204508D-5 |
| Pichia pastoris | ATCC | 28485D-5 |
| Bacillus subtilis | ATCC | 82D-5 |
| Cryptococcus neoformans | ATCC | 208821D-2 |
| Listeria monocytogenes | ATCC | 19114D-5 |
| Staphylococcus aureus | ATCC | 25923D-5 |
| Enterococcus faecalis | ATCC | 700802D-5 |
| ZymoBiomics Microbiome | Zymo Research | D6305 |
| Consumables | Manufacturer | Part Number |
| 384-well PP Microplate | Beckman Coulter Life Sciences | 001-14555 |
| 384-well LDV Plus Microplate | Beckman Coulter Life Sciences | 001-12782 |
| TapeStation Plate | Agilent | #5067-5150 |
| Qubit Microtube | ThermoFisher | #Q32856 |
| 384-well PCR Plate | Bio-Rad | #HSP3805 |
| 384-well Black Flat Clear-bottom Microplate | Greiner | #781096 |
| 1.5 mL DNA LoBind Tubes | Eppendorf | #022431021 |
